「禁忌を含む使用上の注意」等は電子添文をご参照ください
臨床成績 国内第Ⅲ相試験検証試験
(維持血液透析下の二次性副甲状腺機能亢進症患者を対象とした二重盲検並行群間比較試験)[AJ1004試験]
社内資料:第Ⅲ相試験
-維持血液透析下の二次性副甲状腺機能亢進症患者を対象とした二重盲検並行群間比較試験-(承認時評価資料)
試験概要
- 目的
-
維持血液透析下の二次性副甲状腺機能亢進症(SHPT)患者に対するウパシタの有効性について、プラセボを対照とした二重盲検並行群間比較試験にて優越性を検証する。また、安全性についても検討する。
- 対象
-
維持血液透析下のSHPT患者 153例(ウパシタ群103例、プラセボ群50例)
[主な選択基準]週3回の血液透析又は血液透析ろ過を施行する安定期の慢性腎不全患者、連続2週間の最大透析間隔後の透析前血清インタクト副甲状腺ホルモン(iPTH)濃度が240pg/mLを超え、かつ最大透析間隔後の透析前血清補正Ca濃度が8.4mg/dL以上の患者
- 試験デザイン
-
多施設共同、ランダム化、プラセボ対照、二重盲検、並行群間比較
- 方法
-
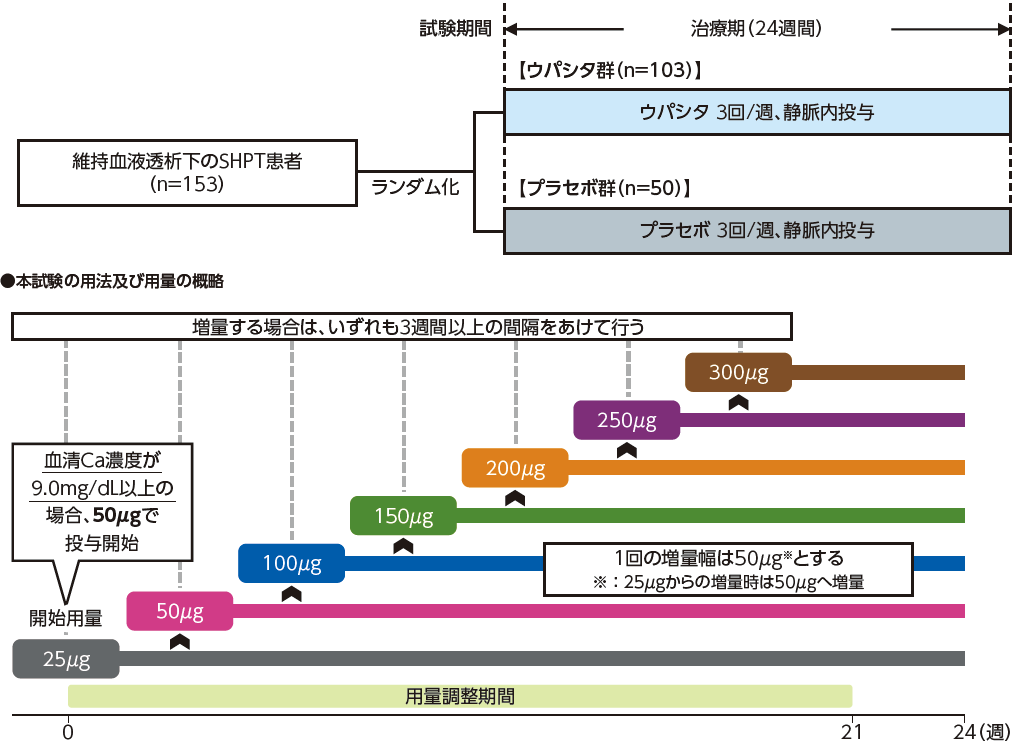
ウパシタ又はプラセボを週3回、血液透析終了時の返血時に透析回路静脈側に用量調整して投与した。期間は24週間とした。
[開始用量]試験薬投与開始1週間前の血清補正Ca濃度が9.0mg/dL未満の場合は25μg、9.0mg/dL以上の場合は50μgとした。
[用量調整方法]患者の血清iPTH濃度を60~240pg/mLの範囲に維持することを目標に、用量を25~300μgの範囲で調整した。なお、用量調整は21週までとし、22~24週までの間は試験薬の用量調整を行わないこととした。ただし、試験薬の減量、休薬及び休薬からの再開は可とした。
[臨床検査]iPTH:スクリーニング時、0~24週は毎週又は中止時の透析前に採血した。
- 評価項目
-
有効性評価項目
<主要評価項目>
22、23、24週における血清iPTH濃度平均値の目標達成率※1
安全性評価項目
有害事象及び副作用、臨床検査値 等
- 解析計画
-
有効性評価項目
有効性評価のための主要解析対象集団は最大の解析対象集団であるFAS(Full Analysis Set)とした。
<主要評価項目>
主要解析として、22、23、24週における血清iPTH濃度平均値が60pg/mL以上240pg/mL以下を達成した患者割合及び両側95%信頼区間を投与群毎に算出した。各投与群の目標達成率について、患者割合の差及び両側95%信頼区間を算出するとともにFisherの直接確率検定を用いて群間比較を行った。
安全性評価項目
副作用について各事象の発現例数及び発現率を算出した。また、注目すべき事象として上部消化管障害、下部消化管障害、低カルシウム血症及び血清補正Ca濃度の低下について、発現例数及び発現率を算出した。
※1:血清iPTH濃度が60pg/mL以上240pg/mL以下を達成した患者の割合
7. 用法及び用量に関連する注意(抜粋)
7.4 増量する場合には増量幅を50μg(ただし25μgから増量する場合は50μgへ増量)とし、2週間以上の間隔をあけて行うこと。
試験デザイン
有効性
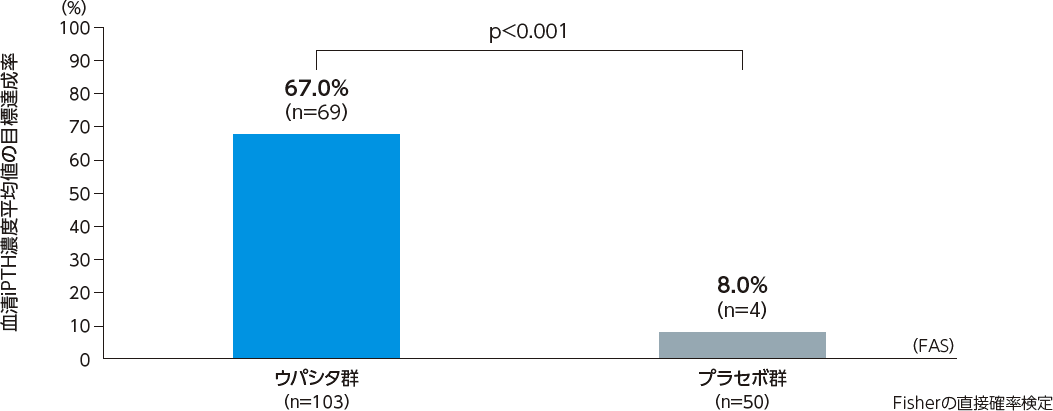
22、23、24週における血清iPTH濃度平均値の目標達成率※[主要評価項目]:検証的解析結果
22、23、24週における血清iPTH濃度平均値の目標達成率は、ウパシタ群で67.0%[95%信頼区間:57.0%-75.9%]、プラセボ群で8.0%[95%信頼区間:2.2%-19.2%]であった。また、投与群間の割合の差は59.0%[95%信頼区間:47.2%-70.8%]であり、プラセボ群に対するウパシタ群の目標達成率は統計学的に有意であることが検証された(p<0.001、Fisherの直接確率検定)。
※:血清iPTH濃度が60pg/mL以上240pg/mL以下を達成した患者の割合
安全性
ウパシタ群では103例中12例(11.7%)、プラセボ群では50例中4例(8.0%)に副作用が認められた。
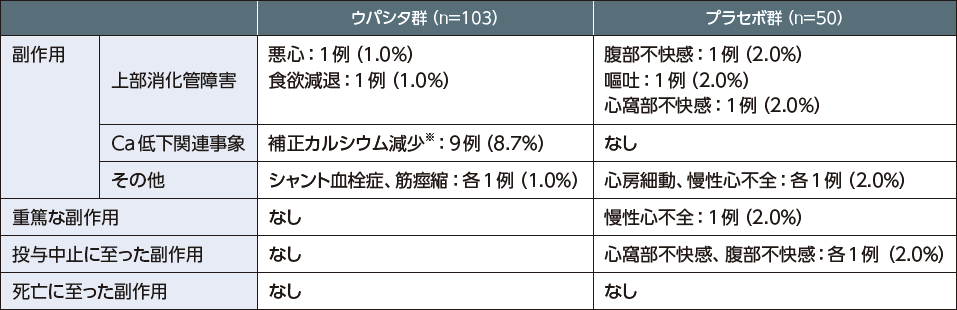
- 集計にはMedDRA/J(バージョン21.1)のPT及びSOCを用いた。
- ※:無症候性のカルシウム減少を「補正カルシウム減少」として集計